Alkaptonuria is a very rare congenital condition. It happens at the time when your body is not able to secrete enough amount of an enzyme known as homogentisic dioxygenase (HGD). This enzyme’s function is to break down a poisonous substance known as homogentisic acid. When your body can’t produce enough HGD, homogentisic acid is accumulated in the body.
This accumulation of homogentisic acid makes your cartilage and bones to turn into faded, discolored and brittle. This usually results in osteoarthritis, particularly in the spine and big joints. Also, those individuals with alkaptonuria have urine that becomes brown or black when it is exposed to air.
In almost half of the AKU cases, the very first symptom comes up at the time of infancy. Medical practitioner and parents might observe dark tints on the diaper. If an infant has alkaptonuria, the homogentisic acid (HGA) in the urine becomes darkened when it is exposed to air.
If the parents and doctors fail to notice the dark urine in childhood, the disease might not be detected until middle age. When the person reaches his 40 the symptoms start to grow. The symptoms grow slowly over the years as the homogentisic acid is accumulated in the body and lay down in cartilage tissue. The most effected body parts are the joints, ears and skin. These organs usually are effected before any other part.
Cartilage is a type connective, tough flexible tissue that could be found in many parts and organs of the body. Cartilage, in fact, shapes the joints between bones, and is responsible for flexibility. HGA built up in the cartilage of joints sooner or later results in arthritis. It most often occur in the spine joints, knees and hips.
Pain and inflexibility in the person’s lower back are usually the very first sings of alkaptonuria.
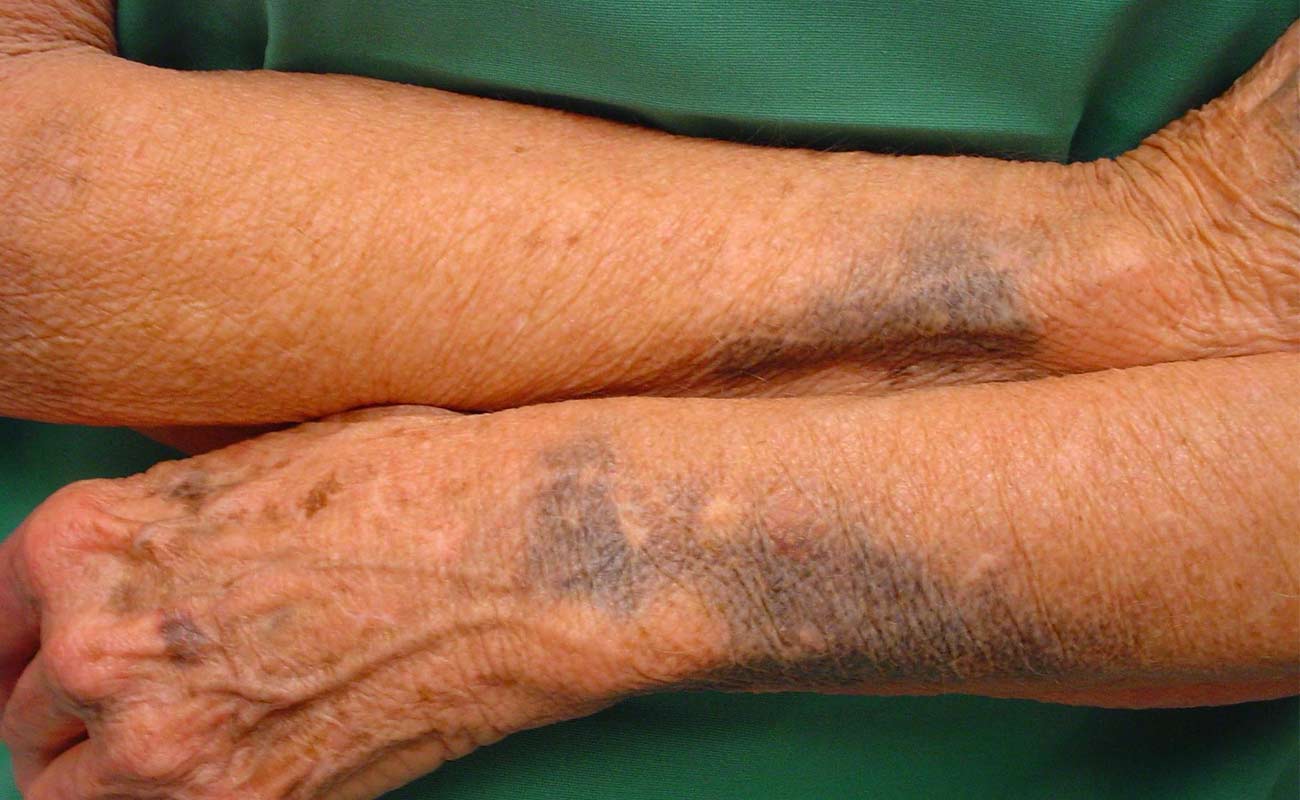
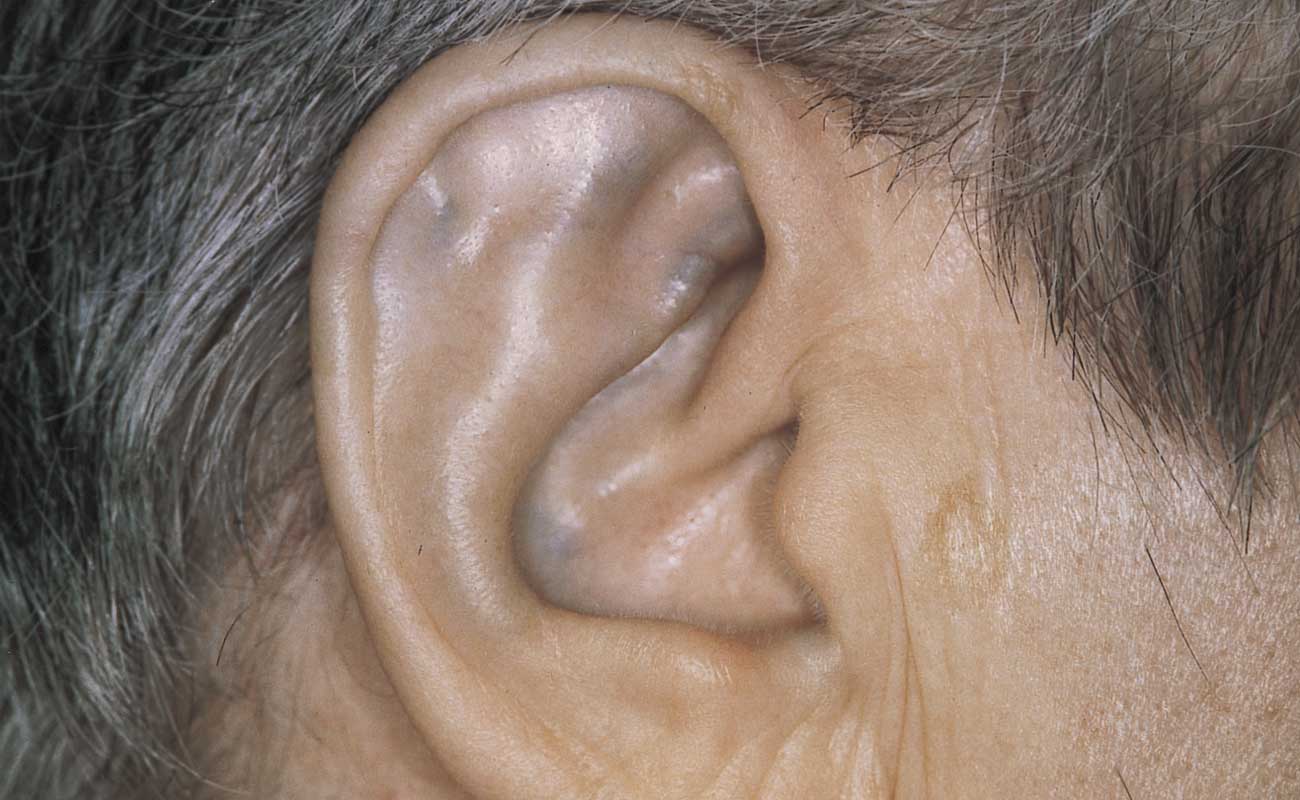
Ear lobe’s flexibility is the result of its cartilage. Since the condition affects the cartilage, an early sign of AKU is thickened and darkened the ear lobes. The ear lobe might turn dark, indicating that HGA is accumulated in ear lobe cartilage. This HGA related discoloration of cartilage is known as ochronosis.
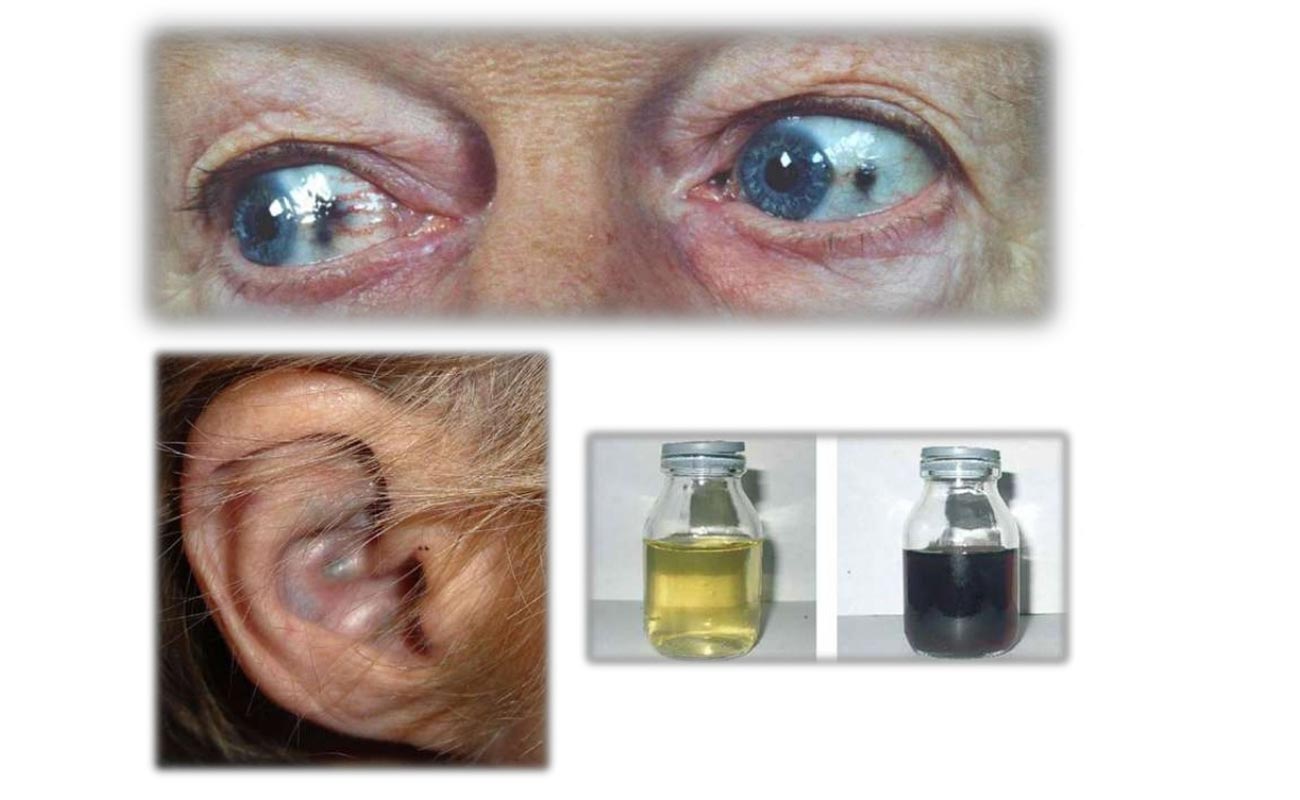
Dark or grey spots may sometimes come up on the cornea or in the sclera (the white section of the eyeball).
Since HGA could be oozed in sweat, after the seat dries out, you may notice some dark spots on your clothes or skin. The dark spots usually can be observed on the forehead and in those areas that are close to sweat glands including the armpits and genital area. Also, the color of the skin under the fingernails can be darkened.
Alkaptonuria Causes
This is a congenital disorder meaning that family history is the main cause of the disease. It, actually, is the only recognized risk factor for Homogentisic Acidura. The leading cause of alkaptonuria is mutations occurred in the HGD gene. The mutation leads to deficiencies in the homogentisate 1, 2-dioxygenase enzyme which impedes the breakdown of tyrosine. Because of these elements, the homogentisic acid, with its oxide called alkapton, begins to build up in the patient’s blood and pass with urine in significant quantities.
Physicians carry out a comprehensive physical exam and ask many questions regarding the symptoms at the moment. The diagnostic exams in this regard include thin layer chromatography and paper chromatography. In order for making diagnosis, the urine as well as blood plasma are employed. Urinalysis and blood tests are crried out to find out if there is any sign of homogentisic acid in the urine. When a person does not have alcaptonuria, Homogentisic acid is not found in his urine and blood plasma but in those with the condition the average urine levels of 3.12 mmol/ mmol of creatinine is found. Also, their average plasma levels are 6.6 micrograms/ ml.
Sometimes, the practitioner add ferric chloride to the urine sample in order to check if it turns black. This is one of the most useful tests for diagnosing alkaptonuria. The other diagnostic procedures that might come in handy in diagnosis include radiology and x-ray. They may be employed for spotting spinal and other abnormalities.
There’s no treatment known for alkaptonuria disorder.
The medical practitioner may recommend a low-protein diet, consuming big amounts of ascorbic acid and vitamin C to decelerate the accumulation of homogentisic acid in the cartilage. Yet, NORD warns that generally the use of vitamin C in long-run has proven to be ineffective for treating alkaptonuria.
Other treatments implemented for alkaptonuria disorder are carried out with concentration on preventing and dismissing other possible complications, including arthritis, heart disease, kidney stones
For instance, the medical professional may prescribe taking anti-inflammatory drugs or tranquillizers to relieve joint pain. Also, physical therapy could be helpful in maintaining flexibility in the muscles and joints. Also, the individual with alkaptonuria should not involve in any activity that put a pressure on his joints.
Some of the patient might need surgery to cope with the consequences of this condition. It is reported that a shoulder, knee, or hip replacement is required for almost half of individuals with alkaptonuria aged 50 or 60. They might also need a surgery to replace their aortic or mitral heart valves. Besides, in some cases, the patient might need surgery or some other therapeutic measures to treat prostate stones or chronic kidney.
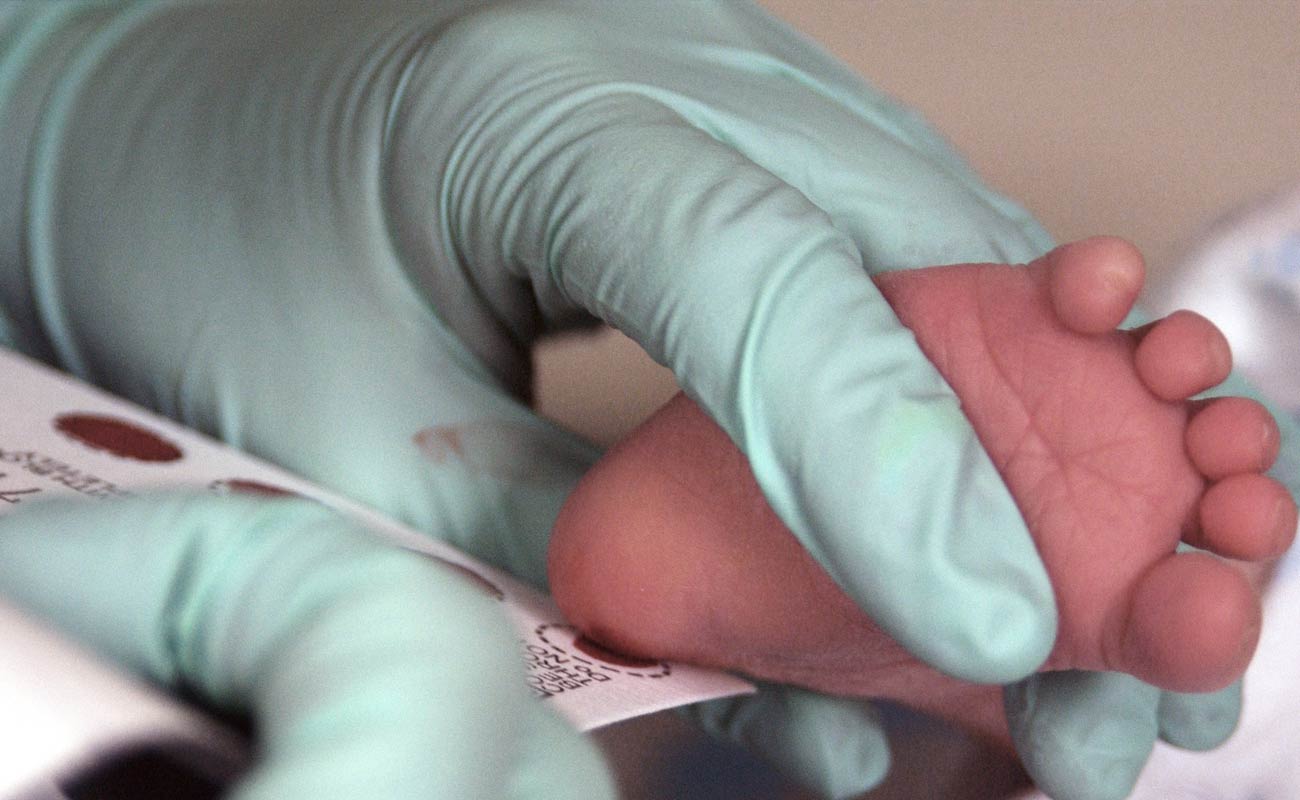
For those with a family history of alkoptonuria who are considering having children, Genetic counseling is recommended.
A blood test may be carried out to find out if one carries the gene for alkaptonuria.
When the genetic alteration is identified, prenatal examinations including amniocentesis or chorionic villus sampling could be conducted to screen a growing infants for this condition.
For further information in this regard, please contact Alale.

Submit Comment